Congratulations to Jacopo Simoni, Gabriele Riva, and Yuan Ping on “First-principles open quantum dynamics for solids based on density-matrix formalism”, published in The Journal of Chemical Physics as a Perspective paper on November 6 2025!
This is a theory for reaching the predictive capability for nonequilibrium quantum many-body systems. Such theory is important to critical fields of QIS, fusion research, microelectronics, and critical materials. Link to the published article.
Abstract:
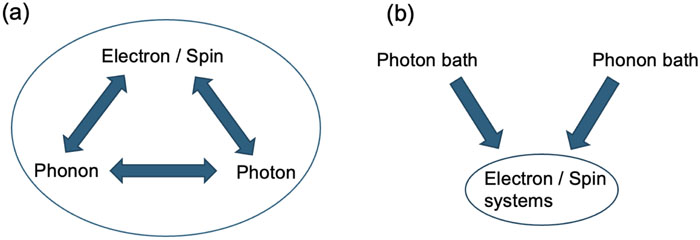
The theoretical description of materials’ properties driven out of equilibrium has important consequences in various fields such as semiconductor spintronics, nonlinear optics, and continuous and discrete quantum information science and technology. The coupling of a quantum many-body system to an external bath can dramatically modify its dynamics compared to that of closed systems; new phenomena like relaxation and decoherence appear as a consequence of the non-unitary evolution of the quantum system. In addition, electron–electron correlations must be properly accounted for in order to go beyond a simple one-electron or mean-field description of the electronic system. Here, we discuss a first-principles methodology based on the evolution of the electronic density matrix capable of treating electron–environment interactions and electron–electron correlations at the same level of description. The effect of the environment is separated into a coherent contribution, like the coupling to applied external electromagnetic fields, and an incoherent contribution, like the interaction with lattice vibrations or the thermal background of radiation. Electron–electron interactions are included using the nonequilibrium Green’s function plus the generalized Kadanoff–Baym ansatz. The obtained non-Markovian coupled set of equations reduces to the ordinary Lindblad quantum master equation form in the Markovian limit.
We acknowledges the support of the DOE Computational Chemical Science program within the Office of Science at DOE under Grant No. DE-SC0023301 and support from the Air Force Office of Scientific Research under Award No. FA9550-21-1-0087.